
Earlier today, I included a paper on the microbiome of pitcher plants, published yesterday in Scientific Reports.
Microbiome and Biocatalytic Bacteria in Monkey Cup (Nepenthes Pitcher) Digestive Fluid – Xin-Yue Cha – Scientific Reports
That’s a really cool topic. Pitcher plants are tropical and carnivorous plants that catch insects and slowly digest them. This paper was studying the digestive fluid isolated from a Malaysian pitcher plant. Figure 1, the only figure of the paper, looked very pretty and colorful at first.
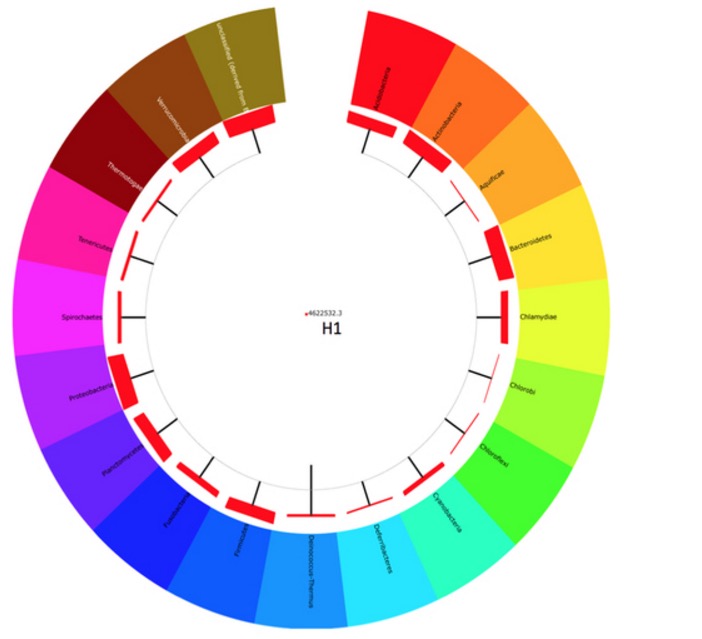
But then I noticed that this was not really a phylogenetic tree. The outer ring shows the phyla found in the pitcher plant, but the lines leading to all these phyla were all directly connected to the same inner line. There was no word in the legend what the thickness of the red boxes meant, but I assume it was denoting the number of reads or OTUs found within each taxon.
The bottom half of Figure 1 was similarly puzzling.
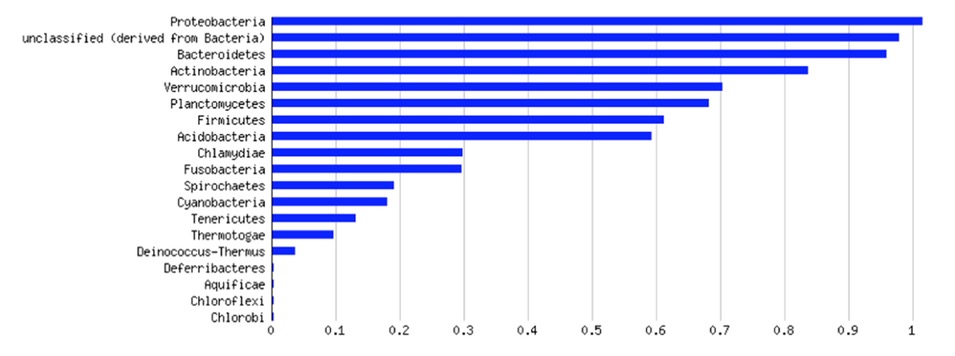
It seems to show that Proteobacteria were the most abundant in this dataset, but it was not clear what the scale or meaning of the X-axis is. The Proteobacteria are a bit of 1.0, so it does not seem to be relative abundance.
I started to read a bit more, and ran into a lot of other unclarities and problems, so many that I even wrote a PubPeer post on it. The paper has language issues, but being a non-native English speaker myself, I did not think that these were the most important problems. Here are some of the problems that I identified during a quick read-through.
Sample collection
Nepenthes pitcher fluid (Sample H1) was collected from wild Nepenthes in Mossy Forest, Pahang, Malaysia (N 04°31′, E 101°22′), at the altitude of 1970 m above sea level. The Nepenthes digestive fluid was transported to laboratory and processed immediately.
According to Google Maps, it would be a 3h car ride from the sampling site to the University of Malaya – were the samples kept on ice or frozen? There is no word on how they samples were transported. Then, how many plants or samples were collected? At some places, the text mentions “sample”, suggesting that only 1 plant/sample was obtained. Did this sample contain visible remnants of trapped insects? Could the microbiome found here be derived from insect guts?
“Targeted Metagenomic Sequencing”
Total DNA extracted from the Nepenthes fluid was subjected to 16S rDNA genes amplification with forward primer (MID1_530F, 5′-ACG AGT GCG TGT GCC AGC MGC NGC GG -3′) and reverse primer (MID1_1100modR, 5′-ACG AGT GCG TGG GTT NCG NTC GTT RC -3′)42. Gene amplification was preformed with gradient annealing temperature from 55 °C to 65 °C. The amplicon sequencing was performed on GS-FLX Titanium platform (Roche, USA).
This paragraph has many problems. First, these primers do not appear to fit on the 16S rRNA gene. I did several BLAST searches, RDP probe match, Google search, but they are not matching to anything. Reference 42, cited here, does not contain these sequences either. It’s a mystery to me what they amplify. Second, this is not metagenomic sequencing; it’s amplifying a specific gene, not analyzing mixed genomes. Then, I was not sure what is meant with the ” gradient annealing temperature” – is this describing a touchdown PCR, or did the authors runn PCRs at multiple annealing temperatures and combined them?
“Taxonomic Assignment of Metagenomic Sequences”
Sequences were trimmed with CLC genomic workbench and annotated with MG-RAST (v3.3) against RDP database (Methods).
A total of 27% of the sequencing read does not match to any DNA sequence in the RDP database. Thus, these reads were listed as “unclassified” (Results)
No word in the methods on quality screening or chimera removal. This is of particular concern since 27% of the 16S rRNA amplicons could not be classified (I assume to phylum level, based on the data shown in Figure 1, but it is not really clear). That is a huge amount of unclassifiable sequences. I wish there is an easy way to quickly download and analyze their 16S rRNA sequence data (SRA SRR916131).
Alpha Diversity

Looking at the plot, the authors found about 600 bacterial species in their sample – I am not sure what is meant by the statement “the alpha diversity of Nepenthes pitcher fluid sample H1 is 31.44”.
Other remarks
The authors did some interesting other analyses, such as culturing (and full-length 16S rRNA gene amplification), MALDI-TOF MS, enzymatic assays, and whole genome sequencing. Those appear to have been executed well, although I am not an expert in some of these techniques. One of the main conclusions, however, seems a bit far-fetched:
The production of a wide range of biocatalytic enzymes by both the Nepenthes and bacteria inhabit in the Nepenthes pitcher digestive fluid contrives a dynamic environment in which both work in synchrony for the decomposition of insects, benefiting both the plant and its microbiota.
Although this might be true, it is a bit of a stretch to base this on the analysis of a single sample from a single plant. Some of the bacteria found in this sample could have been derived from the guts of trapped insects, or just be attached to bark/leaves that fell into the pitcher. This statement would be stronger if the authors had included samples from multiple plants and/or multiple timepoints.
[hr]

Pingback: Blatt is back: “open debate cornerstone of scientific process” – For Better Science